Marker gene identification with COSG#
Setting basics#
[1]:
import numpy as np
import pandas as pd
import scanpy as sc
sc.set_figure_params(dpi=80,dpi_save=300, color_map='Spectral_r',facecolor='white')
from matplotlib import rcParams
# To modify the default figure size, use rcParams.
rcParams['figure.figsize'] = 4, 4
rcParams['font.sans-serif'] = "Arial"
rcParams['font.family'] = "Arial"
sc.settings.verbosity = 3
sc.logging.print_header()
scanpy==1.9.1 anndata==0.8.0 umap==0.5.3 numpy==1.21.6 scipy==1.7.3 pandas==1.4.4 scikit-learn==1.1.2 statsmodels==0.13.2 python-igraph==0.10.4 louvain==0.7.1 pynndescent==0.5.11
Setting colors#
[2]:
### Palette
scane_color=["#e6194b","#3cb44b","#ffe119",
"#4363d8","#f58231","#911eb4",
"#46f0f0","#f032e6","#bcf60c",
"#fabebe","#008080","#e6beff",
"#9a6324",
"#800000",
"#aaffc3","#808000","#ffd8b1",
"#000075","#808080"
]
import seaborn as sns
self_palette2=sns.color_palette(np.hstack((scane_color,sc.pl.palettes.godsnot_102)))
# https://matplotlib.org/3.1.0/tutorials/colors/colormap-manipulation.html#sphx-glr-tutorials-colors-colormap-manipulation-py
# # https://gree2.github.io/python/2015/05/06/python-seaborn-tutorial-choosing-color-palettes
from matplotlib import cm
from matplotlib import colors, colorbar
cmap_own = cm.get_cmap('magma_r', 256)
newcolors = cmap_own(np.linspace(0,0.75 , 256))
Greys = cm.get_cmap('Greys_r', 256)
newcolors[:10, :] = Greys(np.linspace(0.8125, 0.8725, 10))
pos_cmap = colors.ListedColormap(newcolors)
di_cmap=sns.diverging_palette(138,359,s=72,l=50,sep=27,n=15,as_cmap=True)
Setting paths#
[3]:
save_dir='/n/scratch/users/m/mid166/Result/single-cell/Methods/COSG/COSG_tutorial'
sc.settings.figdir = save_dir
prefix='COSG_tutorial_SEA-AD'
import os
if not os.path.exists(save_dir):
os.makedirs(save_dir)
sc.set_figure_params(dpi=80,dpi_save=300, color_map='viridis',facecolor='white')
rcParams['figure.figsize'] = 4, 4
Load the data#
The 20k subsampled snRNA-seq data from Allen SEA-AD project is available in google drive: https://drive.google.com/file/d/1nH-CRaTQFxJ5pAVpy8_hUQn1nrIcakq2/view?usp=drive_link.
The original data is available in https://portal.brain-map.org/explore/seattle-alzheimers-disease.
mkdir -p /n/scratch/users/m/mid166/Result/single-cell/Enhancer/SEA-AD/
cd /n/scratch/users/m/mid166/Result/single-cell/Enhancer/SEA-AD/
gdrive files download 1nH-CRaTQFxJ5pAVpy8_hUQn1nrIcakq2
[4]:
adata=sc.read('/n/scratch/users/m/mid166/Result/single-cell/Enhancer/SEA-AD/SEA-AD_RNA_MTG_subsample_excludeReference_20k_piaso.h5ad')
[5]:
adata.shape
[5]:
(20000, 36601)
[6]:
sc.pl.embedding(adata,
basis='X_umap',
color=['Subclass'],
palette=self_palette2,
legend_fontoutline=2,
legend_fontweight=5,
cmap='Spectral_r',
ncols=3,
size=10,
frameon=False)
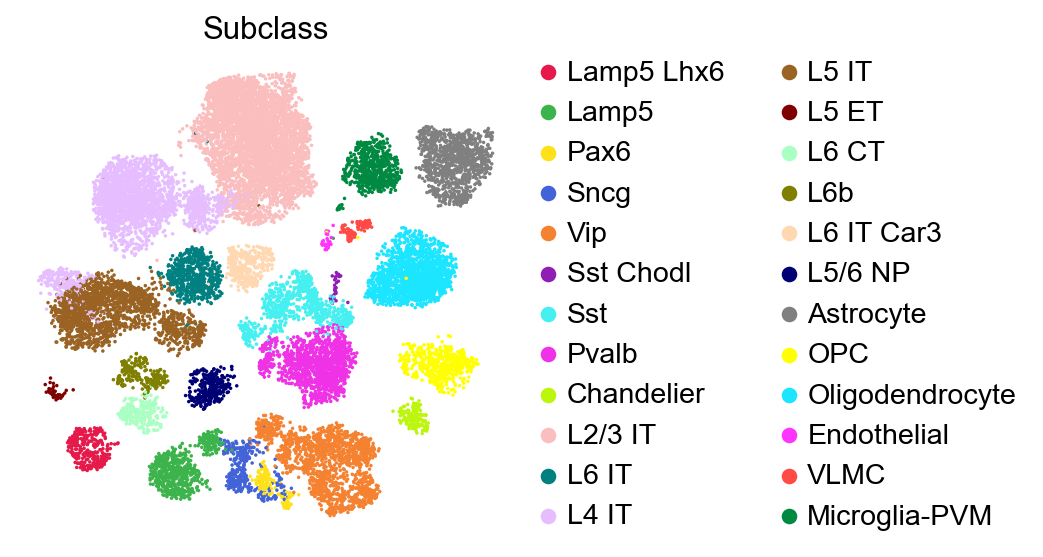
[7]:
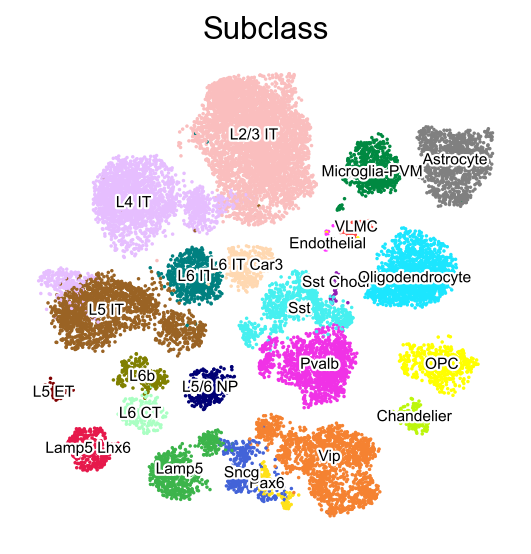
sc.pl.embedding(adata,
basis='X_umap',
color=['Subclass'],
palette=self_palette2,
legend_fontoutline=2,
legend_fontsize=7,
legend_fontweight=5,
legend_loc='on data',
cmap='Spectral_r',
ncols=3,
size=10,
frameon=False)
The data is normalized, and the raw UMI counts were saved in adata.layers['UMIs']:
[8]:
adata.X.data
[8]:
array([0.8813792 , 2.510722 , 0.8813792 , ..., 0.37347588, 0.37347588,
1.1829159 ], dtype=float32)
Import COSG#
Install the latest version of COSG via pip install git+https://github.com/genecell/COSG.git from genecell/COSG:
[9]:
import cosg
[10]:
%%time
groupby='Subclass'
cosg.cosg(adata,
key_added='cosg',
# use_raw=False, layer='log1p', ## e.g., if you want to use the log1p layer in adata
mu=100,
expressed_pct=0.1,
remove_lowly_expressed=True,
n_genes_user=adata.n_vars, ### Use all the genes, to enable the calculation of transformed COSG scores
# n_genes_user=100,
groupby=groupby,
return_by_group=True,
verbosity=1
)
Finished identifying marker genes by COSG, and the results are in adata.uns['cosg'].
CPU times: user 7.79 s, sys: 3.12 s, total: 10.9 s
Wall time: 10.9 s
Let’s visualize the top marker genes identified by COSG with dotplot. There are two ways to build a dendrogram, one with use_rep and one using the category orders in adata.obs[groupby].
Here is the dotplot using the low-dimensional cell representations specified by use_rep:
[11]:
cosg.plotMarkerDotplot(
adata,
groupby='Subclass',
top_n_genes=3,
key_cosg='cosg',
use_rep='X_scVI',
swap_axes=False,
standard_scale='var',
cmap='Spectral_r',
# save='test.pdf'
)
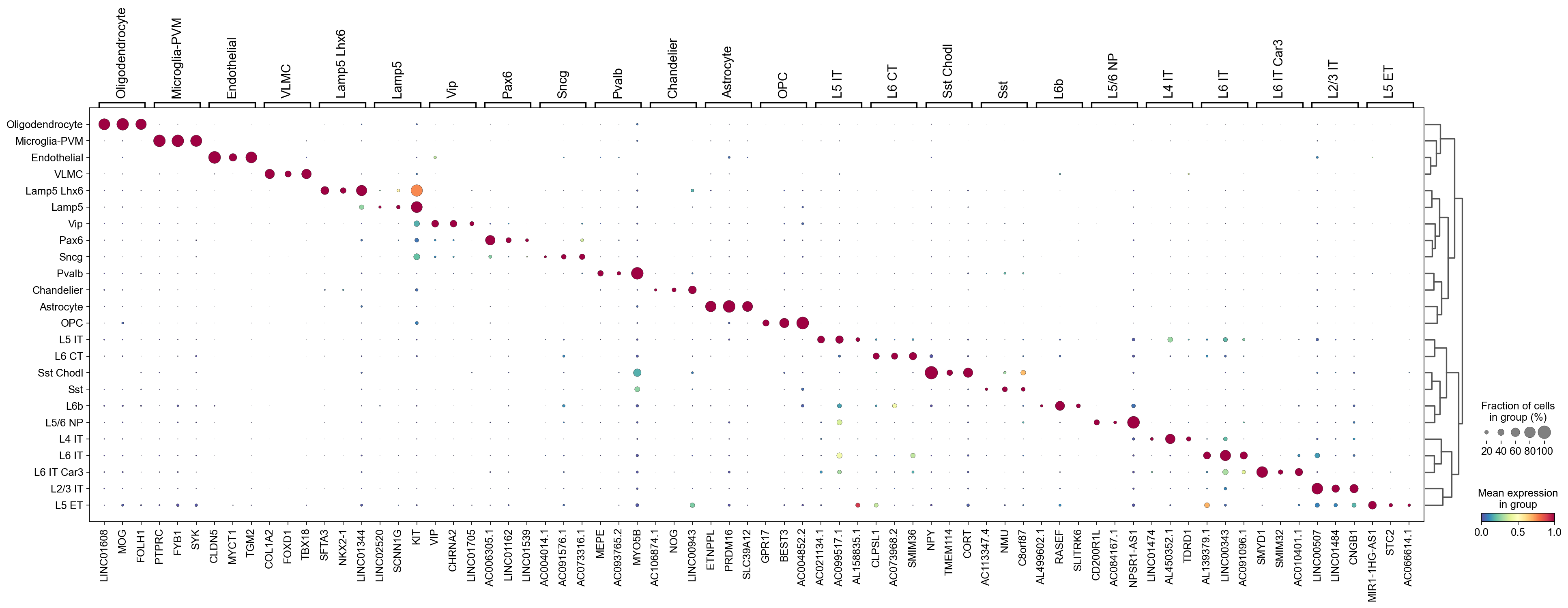
Use the category orders in adata.obs[groupby]:
[12]:
cosg.plotMarkerDotplot(
adata,
groupby='Subclass',
top_n_genes=3,
key_cosg='cosg',
use_rep=None,
swap_axes=False,
standard_scale='var',
cmap='Spectral_r',
# save='test.pdf'
)
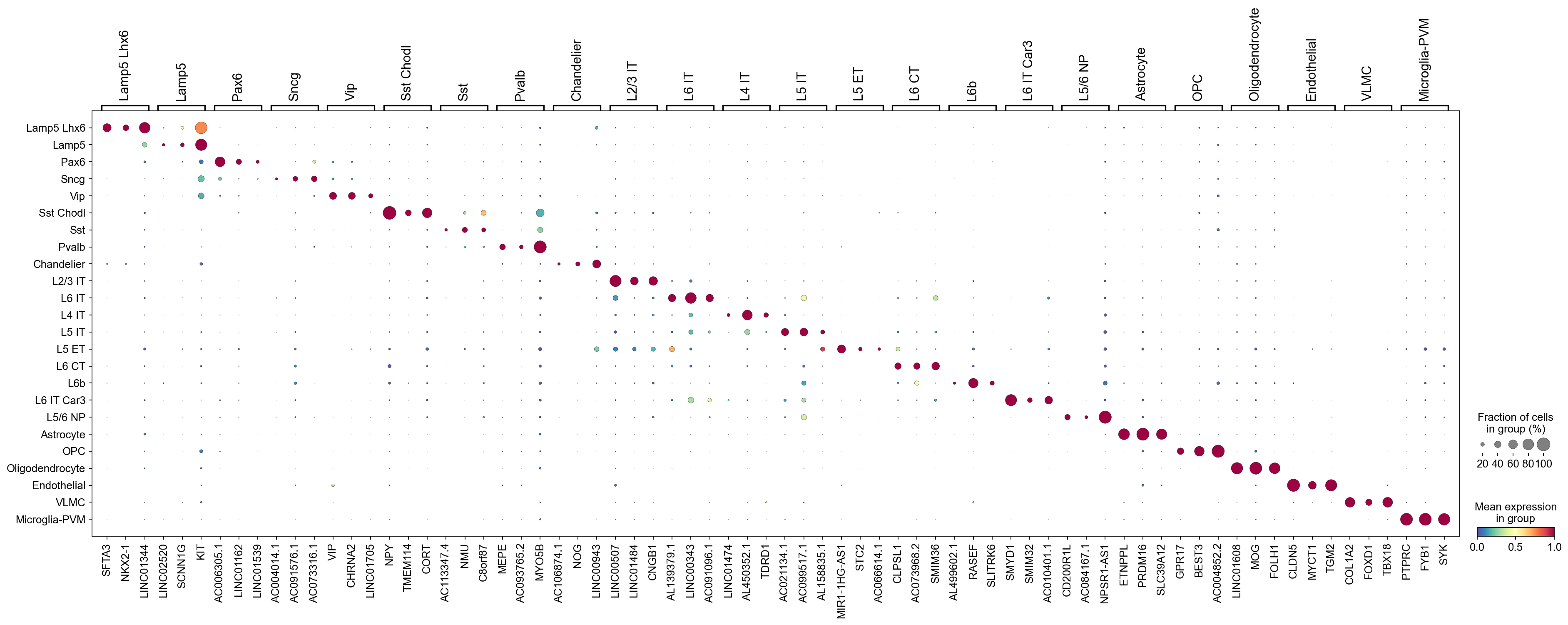
Check the top marker genes:
[13]:
pd.DataFrame(adata.uns['cosg']['names']).head()
[13]:
| Lamp5 Lhx6 | Lamp5 | Pax6 | Sncg | Vip | Sst Chodl | Sst | Pvalb | Chandelier | L2/3 IT | ... | L6 CT | L6b | L6 IT Car3 | L5/6 NP | Astrocyte | OPC | Oligodendrocyte | Endothelial | VLMC | Microglia-PVM | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | SFTA3 | LINC02520 | AC006305.1 | AC004014.1 | VIP | NPY | AC113347.4 | MEPE | AC106874.1 | LINC00507 | ... | CLPSL1 | AL499602.1 | SMYD1 | CD200R1L | ETNPPL | GPR17 | LINC01608 | CLDN5 | COL1A2 | PTPRC |
| 1 | NKX2-1 | SCNN1G | LINC01162 | AC091576.1 | CHRNA2 | TMEM114 | NMU | AC093765.2 | NOG | LINC01484 | ... | AC073968.2 | RASEF | SMIM32 | AC084167.1 | PRDM16 | BEST3 | MOG | MYCT1 | FOXD1 | FYB1 |
| 2 | LINC01344 | KIT | LINC01539 | AC073316.1 | LINC01705 | CORT | C8orf87 | MYO5B | LINC00943 | CNGB1 | ... | SMIM36 | SLITRK6 | AC010401.1 | NPSR1-AS1 | SLC39A12 | AC004852.2 | FOLH1 | TGM2 | TBX18 | SYK |
| 3 | AL355482.1 | AC099482.2 | ABCB5 | AL161629.1 | AJ006995.1 | GHRH | LINC02133 | LRRC38 | AC006065.4 | AC008574.1 | ... | KL | SCUBE1 | AC073225.1 | LINC02042 | OBI1-AS1 | LINC01546 | CD22 | SRARP | C7 | IKZF1 |
| 4 | HCRTR2 | CPLX3 | LINC01416 | AC004862.1 | NPR3 | DDC | OTOF | AC006364.1 | SCUBE3 | LINC02714 | ... | AC092114.1 | FIBCD1 | DCSTAMP | NPSR1 | SLC14A1 | VCAN-AS1 | SLCO1A2 | FENDRR | FMO2 | PIK3R5 |
5 rows × 24 columns
Check the corrsponding COSG scores:
[14]:
pd.DataFrame(adata.uns['cosg']['scores']).head()
[14]:
| Lamp5 Lhx6 | Lamp5 | Pax6 | Sncg | Vip | Sst Chodl | Sst | Pvalb | Chandelier | L2/3 IT | ... | L6 CT | L6b | L6 IT Car3 | L5/6 NP | Astrocyte | OPC | Oligodendrocyte | Endothelial | VLMC | Microglia-PVM | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 0.292173 | 0.058870 | 0.028365 | 0.068951 | 0.350845 | 0.381491 | 0.095953 | 0.362747 | 0.053039 | 0.527400 | ... | 0.013717 | 0.105420 | 0.668806 | 0.431743 | 0.767967 | 0.450832 | 0.832948 | 0.801609 | 0.763216 | 0.871644 |
| 1 | 0.231243 | 0.019967 | 0.012673 | 0.068790 | 0.328894 | 0.053314 | 0.049051 | 0.172069 | 0.028827 | 0.482379 | ... | 0.009300 | 0.068174 | 0.173390 | 0.327094 | 0.763356 | 0.419320 | 0.806407 | 0.573506 | 0.584916 | 0.856651 |
| 2 | 0.026953 | 0.017913 | 0.006920 | 0.019831 | 0.268984 | 0.011365 | 0.021399 | 0.126399 | 0.024365 | 0.317597 | ... | 0.006055 | 0.067735 | 0.093702 | 0.283468 | 0.725767 | 0.351603 | 0.799328 | 0.571553 | 0.583440 | 0.834092 |
| 3 | 0.013707 | 0.016702 | 0.003892 | 0.016700 | 0.262942 | 0.008558 | 0.020437 | 0.117930 | 0.022924 | 0.264376 | ... | 0.004044 | 0.040743 | 0.092139 | 0.157058 | 0.698517 | 0.310609 | 0.772911 | 0.567358 | 0.469173 | 0.822119 |
| 4 | 0.011856 | 0.015210 | 0.003594 | 0.012816 | 0.254974 | 0.007424 | 0.013169 | 0.033898 | 0.020663 | 0.204915 | ... | 0.003518 | 0.025265 | 0.068585 | 0.111516 | 0.687068 | 0.297090 | 0.761675 | 0.554236 | 0.429890 | 0.819302 |
5 rows × 24 columns
The COSG results are also saved as a multi-index Pandas dataframe format if you set return_by_group=True (by default), and you can access it via:
[15]:
adata.uns['cosg']['COSG'].head()
[15]:
| names::Lamp5 Lhx6 | scores::Lamp5 Lhx6 | names::Lamp5 | scores::Lamp5 | names::Pax6 | scores::Pax6 | names::Sncg | scores::Sncg | names::Vip | scores::Vip | ... | names::OPC | scores::OPC | names::Oligodendrocyte | scores::Oligodendrocyte | names::Endothelial | scores::Endothelial | names::VLMC | scores::VLMC | names::Microglia-PVM | scores::Microglia-PVM | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | SFTA3 | 0.292173 | LINC02520 | 0.058870 | AC006305.1 | 0.028365 | AC004014.1 | 0.068951 | VIP | 0.350845 | ... | GPR17 | 0.450832 | LINC01608 | 0.832948 | CLDN5 | 0.801609 | COL1A2 | 0.763216 | PTPRC | 0.871644 |
| 1 | NKX2-1 | 0.231243 | SCNN1G | 0.019967 | LINC01162 | 0.012673 | AC091576.1 | 0.068790 | CHRNA2 | 0.328894 | ... | BEST3 | 0.419320 | MOG | 0.806407 | MYCT1 | 0.573506 | FOXD1 | 0.584916 | FYB1 | 0.856651 |
| 2 | LINC01344 | 0.026953 | KIT | 0.017913 | LINC01539 | 0.006920 | AC073316.1 | 0.019831 | LINC01705 | 0.268984 | ... | AC004852.2 | 0.351603 | FOLH1 | 0.799328 | TGM2 | 0.571553 | TBX18 | 0.583440 | SYK | 0.834092 |
| 3 | AL355482.1 | 0.013707 | AC099482.2 | 0.016702 | ABCB5 | 0.003892 | AL161629.1 | 0.016700 | AJ006995.1 | 0.262942 | ... | LINC01546 | 0.310609 | CD22 | 0.772911 | SRARP | 0.567358 | C7 | 0.469173 | IKZF1 | 0.822119 |
| 4 | HCRTR2 | 0.011856 | CPLX3 | 0.015210 | LINC01416 | 0.003594 | AC004862.1 | 0.012816 | NPR3 | 0.254974 | ... | VCAN-AS1 | 0.297090 | SLCO1A2 | 0.761675 | FENDRR | 0.554236 | FMO2 | 0.429890 | PIK3R5 | 0.819302 |
5 rows × 48 columns
Calculate normalized COSG scores for each gene across all cell types#
In addition to COSG scores, the normalized COSG scores (or the transformed COSG scores) can also be calculated to enable cross-cell type comparison of the gene expression specificities. This could also potentially enables the expression specificity comparison across datasets or stuides.
Note: setting n_genes_user = adata.n_vars in the cosg.cosg() function is required for calculation of normalized COSG scores.
We firstly reindex the marker gene dataframes by indexing them with the same gene indexes, and the values are the COSG scores in each cell type:
[16]:
cosg_scores=cosg.indexByGene(
adata.uns['cosg']['COSG'],
# gene_key="names", score_key="scores",
set_nan_to_zero=True,
convert_negative_one_to_zero=True
)
[17]:
cosg_scores.head()
[17]:
| Lamp5 Lhx6 | Lamp5 | Pax6 | Sncg | Vip | Sst Chodl | Sst | Pvalb | Chandelier | L2/3 IT | ... | L6 CT | L6b | L6 IT Car3 | L5/6 NP | Astrocyte | OPC | Oligodendrocyte | Endothelial | VLMC | Microglia-PVM | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SFTA3 | 0.292173 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.000000 | ... | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| NKX2-1 | 0.231243 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.000000 | ... | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| LINC01344 | 0.026953 | 0.000317 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.000000 | ... | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| AL355482.1 | 0.013707 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.000000 | ... | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| HCRTR2 | 0.011856 | 0.000000 | 0.000058 | 0.000023 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.000028 | ... | 0.000035 | 0.000153 | 0.000002 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
5 rows × 24 columns
Next, we normalize the COSG scores:
[18]:
cosg_scores=cosg.iqrLogNormalize(cosg_scores)
[19]:
cosg_scores.head()
[19]:
| Lamp5 Lhx6 | Lamp5 | Pax6 | Sncg | Vip | Sst Chodl | Sst | Pvalb | Chandelier | L2/3 IT | ... | L6 CT | L6b | L6 IT Car3 | L5/6 NP | Astrocyte | OPC | Oligodendrocyte | Endothelial | VLMC | Microglia-PVM | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SFTA3 | 9.762279 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.000000 | ... | 0.000000 | 0.000000 | 0.00000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| NKX2-1 | 9.528420 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.000000 | ... | 0.000000 | 0.000000 | 0.00000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| LINC01344 | 7.379579 | 2.019498 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.000000 | ... | 0.000000 | 0.000000 | 0.00000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| AL355482.1 | 6.703997 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.000000 | ... | 0.000000 | 0.000000 | 0.00000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| HCRTR2 | 6.559136 | 0.000000 | 2.468799 | 0.862006 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.017969 | ... | 1.405817 | 2.548247 | 0.05476 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
5 rows × 24 columns
Let’s check some genes’ normalized scores:
[20]:
gene_check=np.ravel(pd.DataFrame(adata.uns['cosg']['names'])[:1].values.T)[:5]
[21]:
cosg_scores.loc[gene_check].T.head()
[21]:
| SFTA3 | LINC02520 | AC006305.1 | AC004014.1 | VIP | |
|---|---|---|---|---|---|
| Lamp5 Lhx6 | 9.762279 | 0.000000 | 0.000000 | 0.000000 | 0.00000 |
| Lamp5 | 0.000000 | 7.103147 | 0.000000 | 0.000000 | 0.00000 |
| Pax6 | 0.000000 | 0.000000 | 8.580213 | 0.000000 | 0.00000 |
| Sncg | 0.000000 | 0.000000 | 2.847808 | 8.320658 | 0.00000 |
| Vip | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 7.59415 |
and here are the COSG scores before normalization, which are not directly comparable across cell types, as they are influenced by the cell numbers and cell cluster similarities, etc.
[22]:
cosg_scores_original=cosg.indexByGene(
adata.uns['cosg']['COSG'],
# gene_key="names", score_key="scores",
set_nan_to_zero=True,
convert_negative_one_to_zero=True
)
cosg_scores_original.loc[gene_check].T.head()
[22]:
| SFTA3 | LINC02520 | AC006305.1 | AC004014.1 | VIP | |
|---|---|---|---|---|---|
| Lamp5 Lhx6 | 0.292173 | 0.00000 | 0.000000 | 0.000000 | 0.000000 |
| Lamp5 | 0.000000 | 0.05887 | 0.000000 | 0.000000 | 0.000000 |
| Pax6 | 0.000000 | 0.00000 | 0.028365 | 0.000000 | 0.000000 |
| Sncg | 0.000000 | 0.00000 | 0.000273 | 0.068951 | 0.000000 |
| Vip | 0.000000 | 0.00000 | 0.000000 | 0.000000 | 0.350845 |
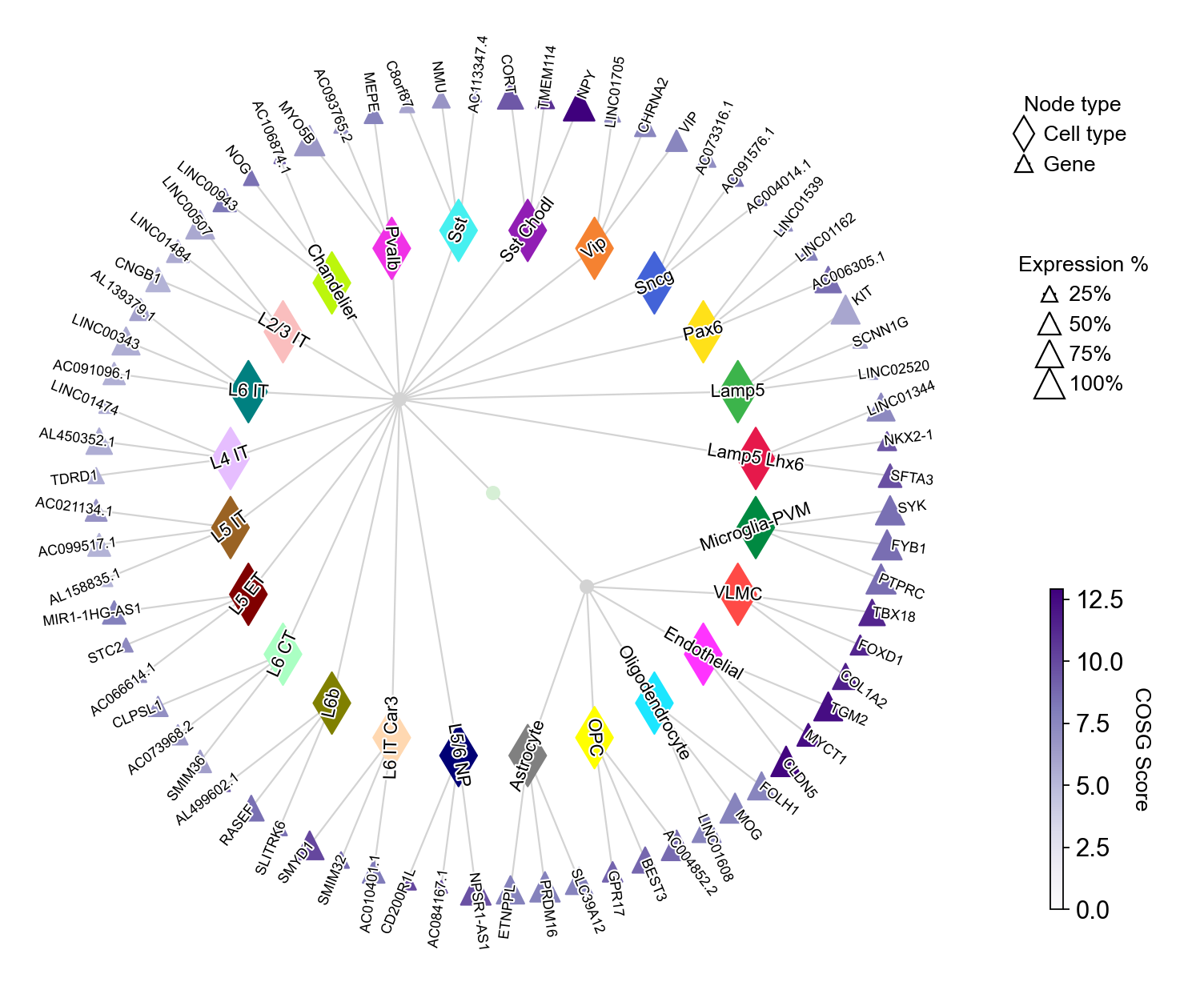
Visualizing the marker genes with plotMarkerDendrogram#
COSG provides an intutive visualization with marker gene names, COSG scores and cell cluster similarities:
[23]:
cosg.plotMarkerDendrogram(
adata,
group_by="Subclass",
use_rep="X_scVI",
calculate_dendrogram_on_cosg_scores=False,
top_n_genes=3,
radius_step=1.5,
cmap="Purples",
gene_label_offset=0.25,
gene_label_color="black",
linkage_method="ward",
distance_metric="correlation",
hierarchy_merge_scale=0,
# collapse_scale=0.2,
add_cluster_node_for_single_node_cluster=True,
palette=None,
figure_size= (10, 10),
# node_shape_cell_type='o',
# node_shape_gene='s',
colorbar_width=0.01,
gene_color_min=0,
gene_color_max=None,
show_figure=True,
# save='./markerDendrogram.pdf',
)
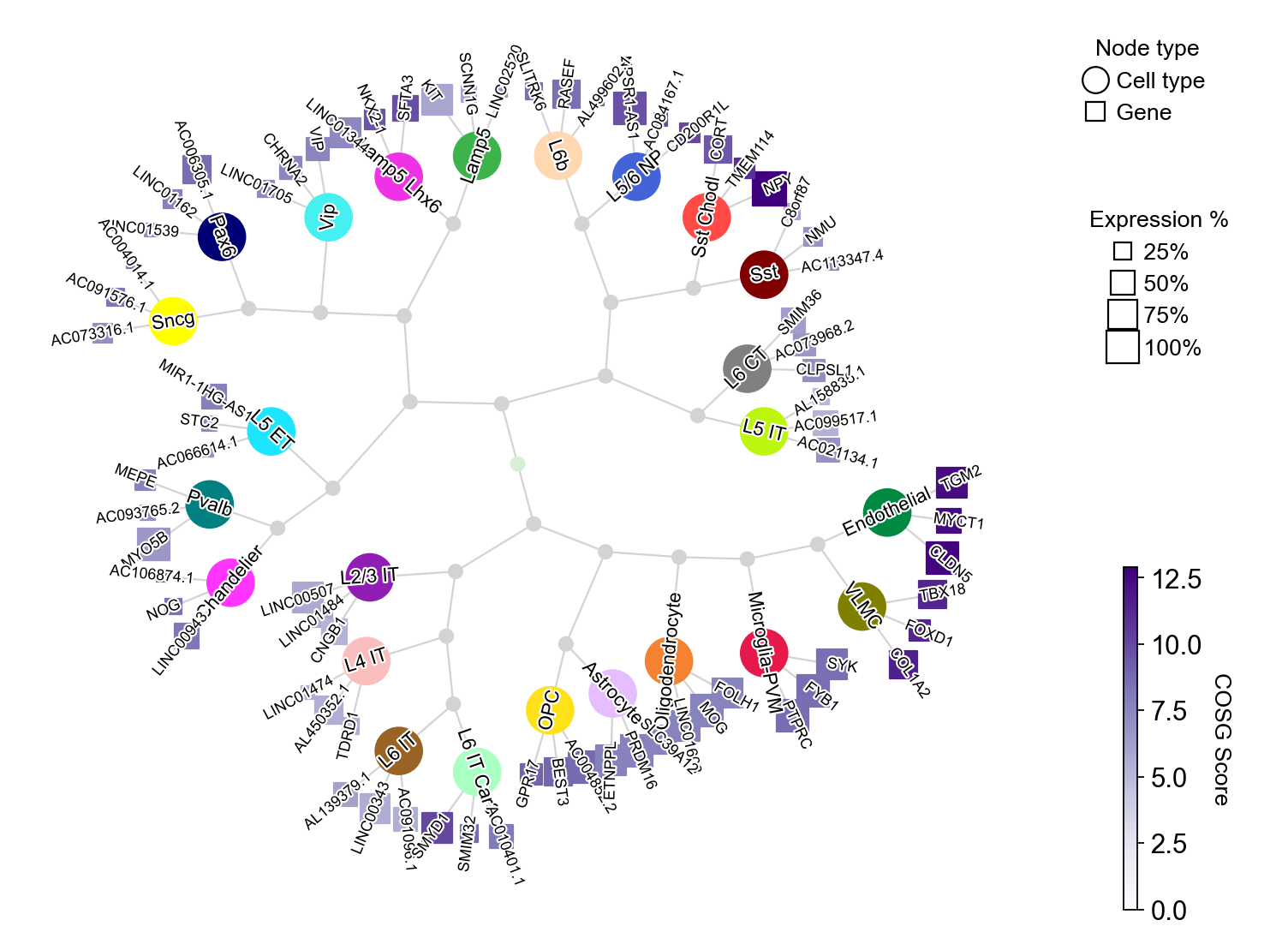
You could set the gene node colors by cmap and merge the hierarchical dendrogram by hierarchy_merge_scale. The parameter hierarchy_merge_scale (0 to 1) controls merging binary nodes into multi-child nodes based on distance similarity:
[24]:
cosg.plotMarkerDendrogram(
adata,
group_by="Subclass",
use_rep="X_scVI",
calculate_dendrogram_on_cosg_scores=True,
top_n_genes=3,
radius_step=1.5,
cmap="Spectral_r",
gene_label_offset=0.25,
gene_label_color="black",
linkage_method="ward",
distance_metric="correlation",
hierarchy_merge_scale=0.2,
# collapse_scale=0.2,
add_cluster_node_for_single_node_cluster=True,
palette=None,
figure_size= (10, 10),
# figure_size= (8, 8),
# node_shape_cell_type='o',
# node_shape_gene='s',
colorbar_width=0.01,
gene_color_min=0,
gene_color_max=None,
show_figure=True,
# save='./markerDendrogram.pdf',
)
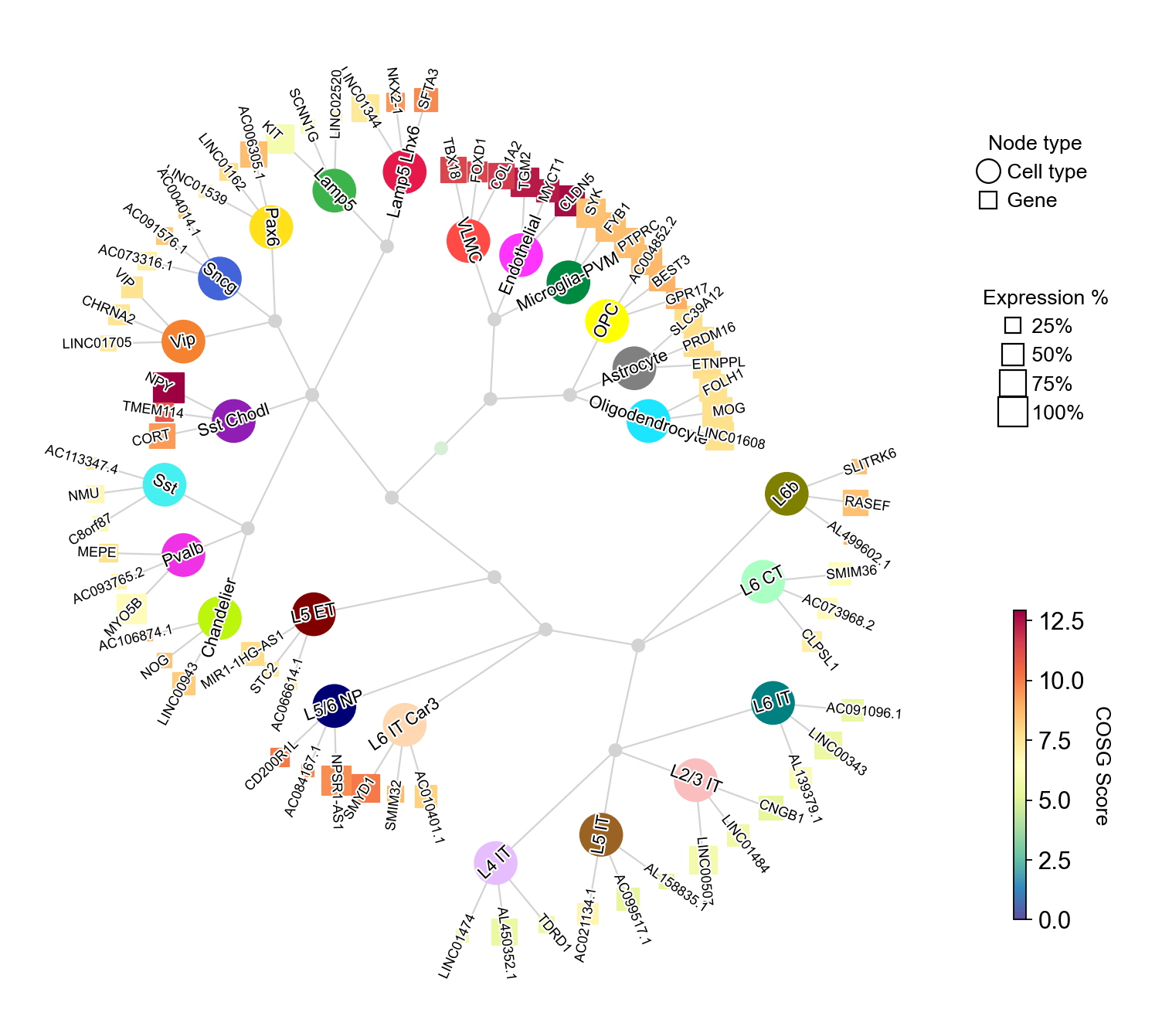
You could also collapse the dendrogram by setting collapse_scale to a value between 0 and 1:
[25]:
cosg.plotMarkerDendrogram(
adata,
group_by="Subclass",
use_rep="X_scVI",
calculate_dendrogram_on_cosg_scores=True,
top_n_genes=3,
radius_step=1.5,
cmap="Purples",
gene_label_offset=0.25,
gene_label_color="black",
linkage_method="ward",
distance_metric="correlation",
hierarchy_merge_scale=0,
collapse_scale=0.2,
add_cluster_node_for_single_node_cluster=True,
palette=None,
figure_size= (10, 10),
# node_shape_cell_type='o',
# node_shape_gene='s',
colorbar_width=0.01,
gene_color_min=0,
gene_color_max=None,
show_figure=True,
# save='./markerDendrogram.pdf',
)
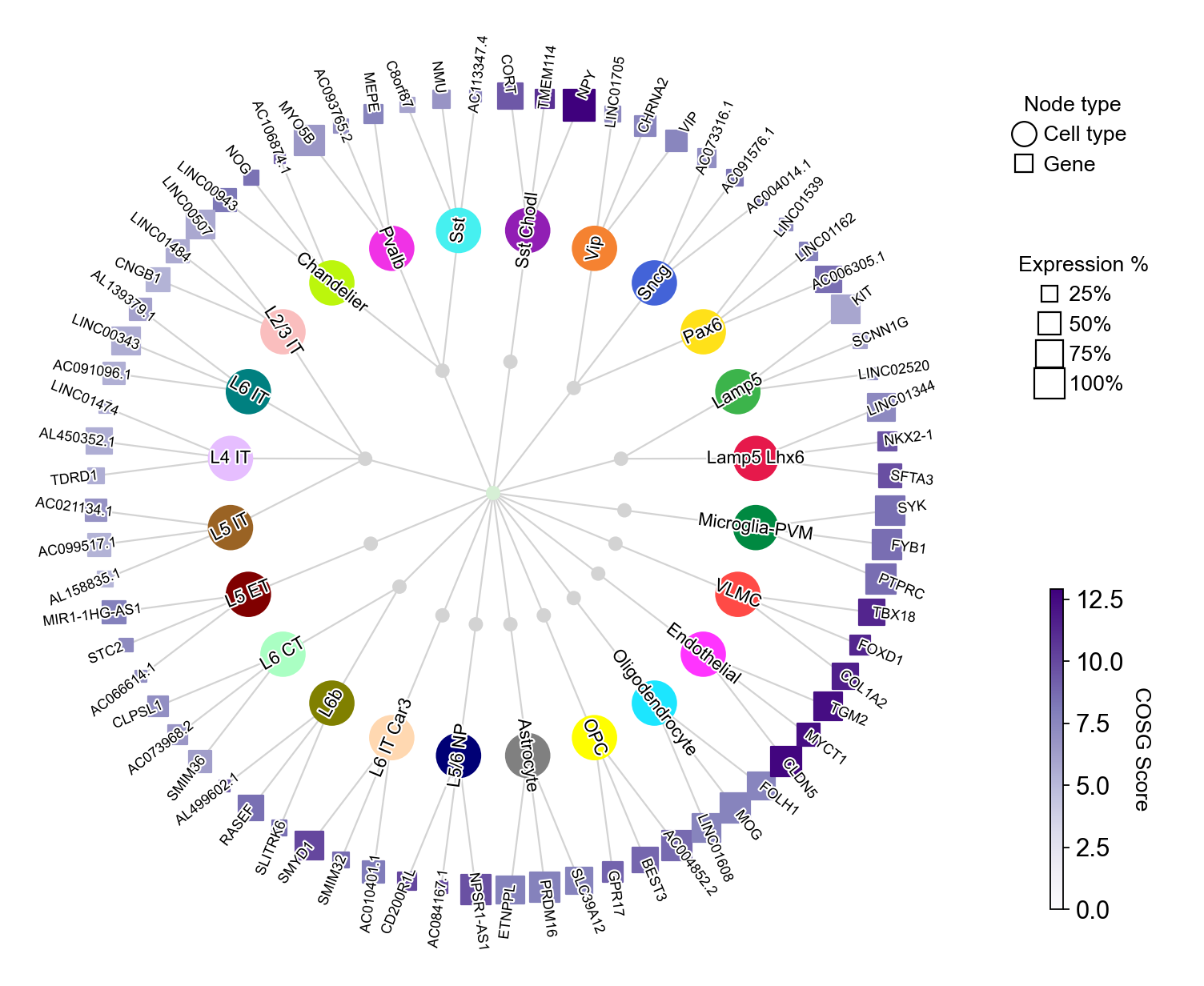
The parameter collapse_scale controls the clustering resolution of cell types, larger values lead to more clusters:
[26]:
cosg.plotMarkerDendrogram(
adata,
group_by="Subclass",
use_rep="X_scVI",
calculate_dendrogram_on_cosg_scores=True,
top_n_genes=3,
radius_step=1.5,
cmap="Purples",
gene_label_offset=0.25,
gene_label_color="black",
linkage_method="ward",
distance_metric="correlation",
hierarchy_merge_scale=0,
collapse_scale=0.8,
add_cluster_node_for_single_node_cluster=True,
palette=None,
figure_size= (10, 10),
# node_shape_cell_type='o',
# node_shape_gene='s',
colorbar_width=0.01,
gene_color_min=0,
gene_color_max=None,
show_figure=True,
# save='./markerDendrogram.pdf',
)
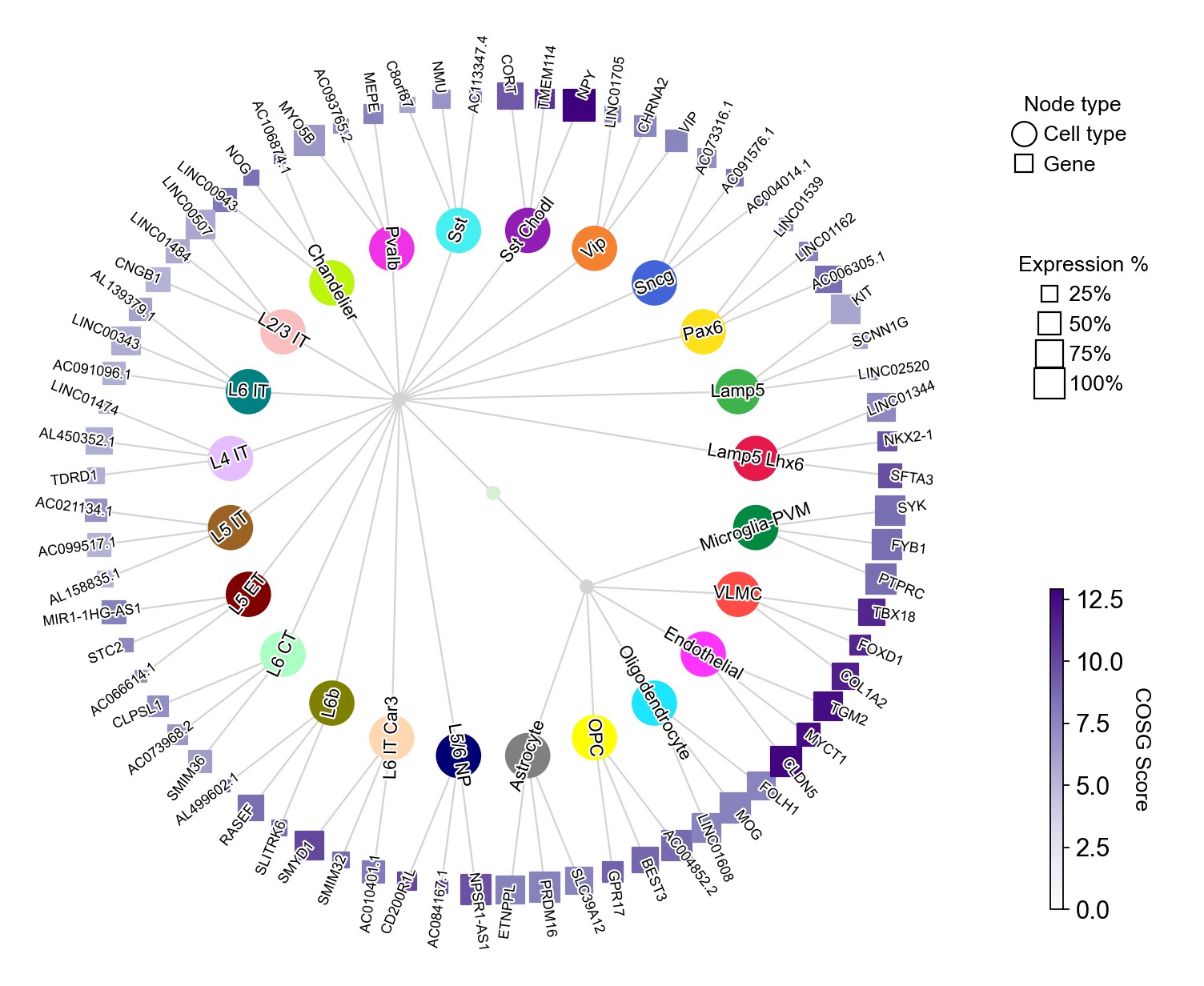
Besides, you could also not show the figure by setting show_figure=False and save the figure as a PDF file by save='./markerDendrogram.pdf':
[27]:
cosg.plotMarkerDendrogram(
adata,
group_by="Subclass",
use_rep="X_scVI",
calculate_dendrogram_on_cosg_scores=True,
top_n_genes=3,
radius_step=1.5,
cmap="Purples",
gene_label_offset=0.25,
gene_label_color="black",
linkage_method="ward",
distance_metric="correlation",
hierarchy_merge_scale=0,
collapse_scale=0.2,
add_cluster_node_for_single_node_cluster=True,
palette=None,
figure_size= (10, 10),
# node_shape_cell_type='o',
# node_shape_gene='s',
colorbar_width=0.01,
gene_color_min=0,
gene_color_max=None,
show_figure=False,
# save='./markerDendrogram.pdf',
)
The shape of cell type nodes and gene nodes could be adjusted via node_shape_cell_type and node_shape_gene, respectively:
[28]:
cosg.plotMarkerDendrogram(
adata,
group_by="Subclass",
use_rep="X_scVI",
calculate_dendrogram_on_cosg_scores=True,
top_n_genes=3,
radius_step=1.5,
cmap="Purples",
gene_label_offset=0.25,
gene_label_color="black",
linkage_method="ward",
distance_metric="correlation",
hierarchy_merge_scale=0,
collapse_scale=0.8,
add_cluster_node_for_single_node_cluster=True,
palette=None,
figure_size= (10, 10),
node_shape_cell_type='d',
node_shape_gene='^',
colorbar_width=0.01,
gene_color_min=0,
gene_color_max=None,
show_figure=True,
# save='./markerDendrogram.pdf',
)
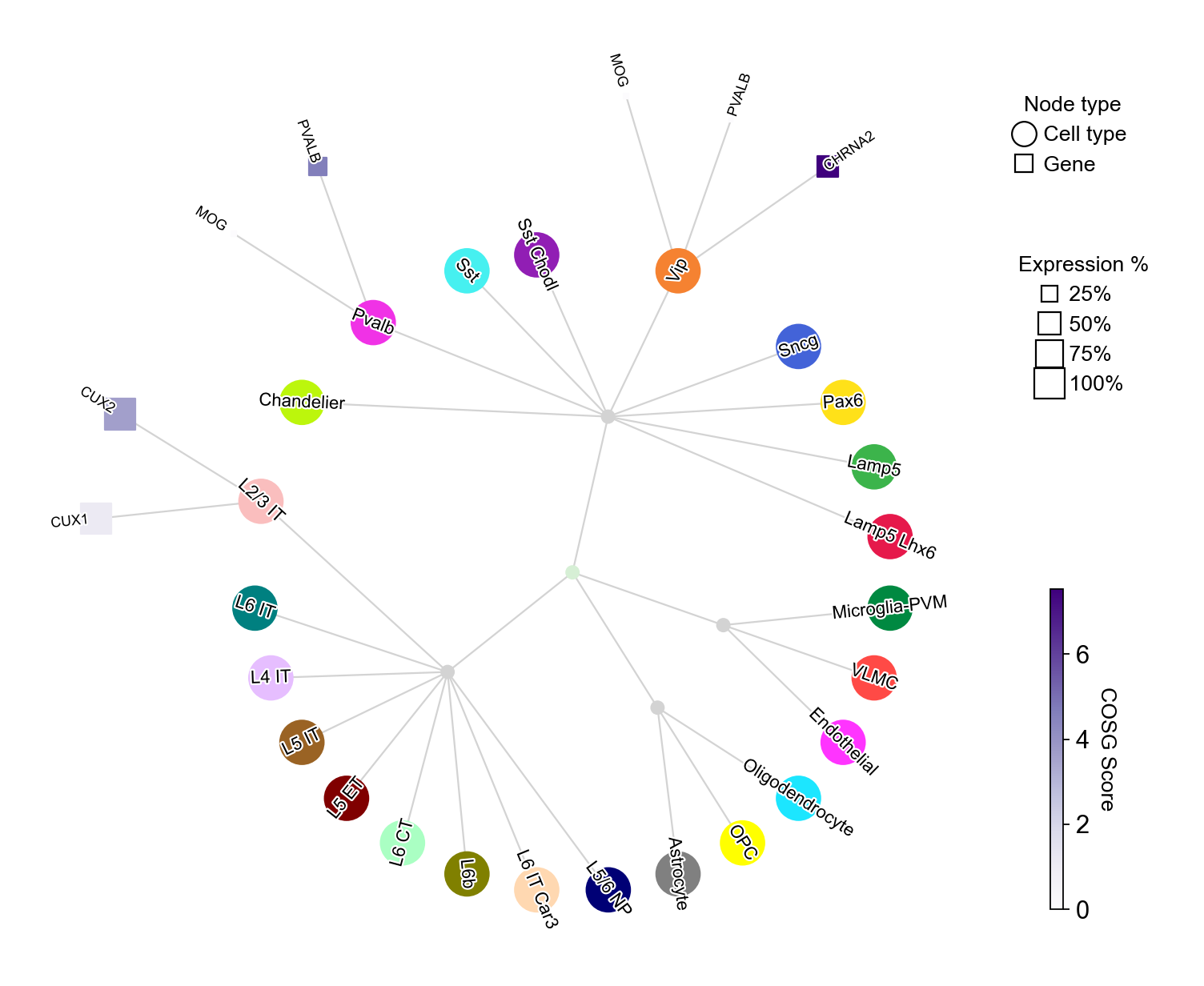
Use customized cell type-gene pairs for visualization#
[29]:
celltype_gene_mapping={
'L2/3 IT': ['CUX2', 'CUX1'],
'Pvalb': ['PVALB', 'MOG', ],
'Vip': ['CHRNA2', 'PVALB', 'MOG', ],
}
[30]:
cosg.plotMarkerDendrogram(
adata,
group_by="Subclass",
use_rep="X_scVI",
calculate_dendrogram_on_cosg_scores=True,
top_n_genes=3,
map_cell_type_gene=celltype_gene_mapping,
cell_type_selected=None,
radius_step=1.5,
# cmap="Spectral_r",
# cmap="magma_r",
cmap="Purples",
gene_label_offset=0.25,
gene_label_color="black",
linkage_method="ward",
distance_metric="correlation",
# hierarchy_merge_scale=0.2,
collapse_scale=0.5,
add_cluster_node_for_single_node_cluster=False,
palette=None,
figure_size= (10, 10),
# node_shape_cell_type='o',
# node_shape_gene='s',
colorbar_width=0.01,
gene_color_min=0,
gene_color_max=None,
# edge_curved=0.5,
show_figure=True,
# save='./markerDendrogram.pdf',
)
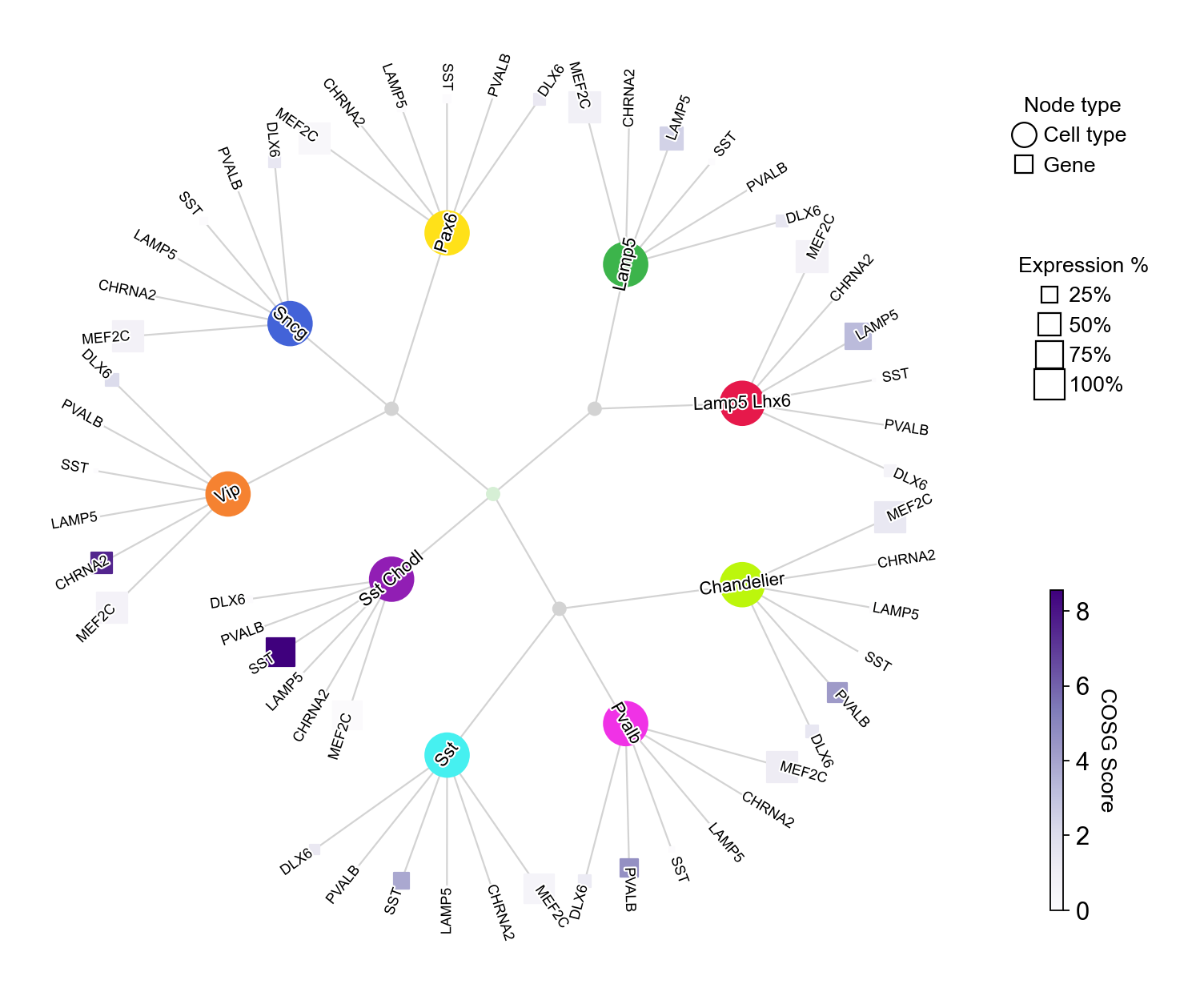
We could also combine the cell type filtering via cell_type_selected with customized cell type-gene pairs:
[31]:
celltype_gene_mapping={cell_type: ['DLX6', 'PVALB', 'SST', 'LAMP5', 'CHRNA2', 'MEF2C' ] for cell_type in adata.obs['Subclass'].cat.categories}
[32]:
cosg.plotMarkerDendrogram(
adata,
group_by="Subclass",
use_rep="X_scVI",
calculate_dendrogram_on_cosg_scores=True,
top_n_genes=3,
map_cell_type_gene=celltype_gene_mapping,
cell_type_selected=['Lamp5 Lhx6', 'Lamp5', 'Pax6', 'Sncg', 'Vip', 'Sst Chodl', 'Sst',
'Pvalb', 'Chandelier',],
radius_step=1.5,
# cmap="Spectral_r",
# cmap="magma_r",
cmap="Purples",
gene_label_offset=0.25,
gene_label_color="black",
linkage_method="ward",
distance_metric="correlation",
# hierarchy_merge_scale=0.2,
collapse_scale=0.5,
add_cluster_node_for_single_node_cluster=False,
palette=None,
figure_size= (10, 10),
# node_shape_cell_type='o',
# node_shape_gene='s',
colorbar_width=0.01,
gene_color_min=0,
gene_color_max=None,
# edge_curved=0.5,
show_figure=True,
# save='./markerDendrogram.pdf',
)
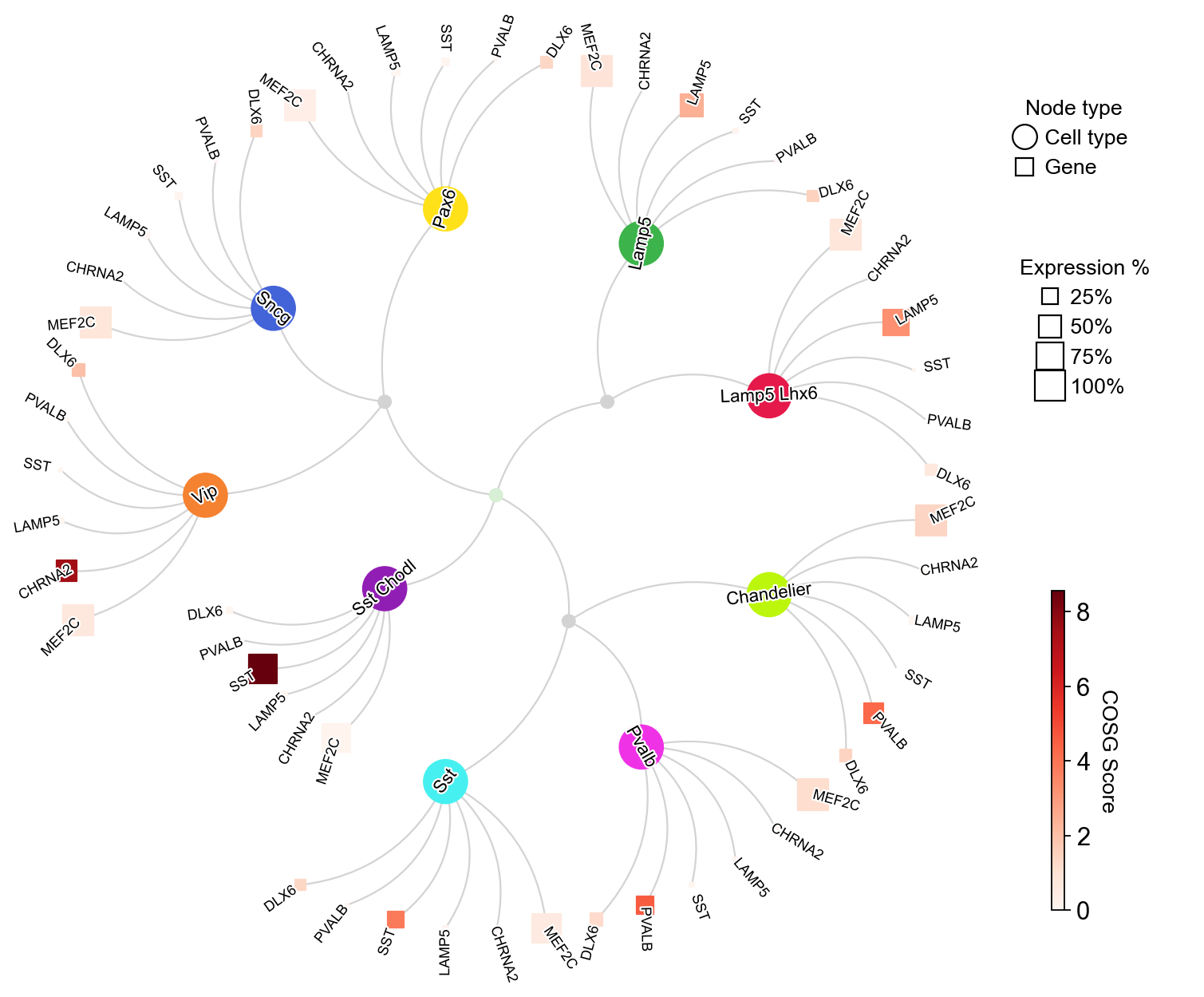
We can also change the curvature of edges via edge_curved:
[33]:
cosg.plotMarkerDendrogram(
adata,
group_by="Subclass",
use_rep="X_scVI",
calculate_dendrogram_on_cosg_scores=True,
top_n_genes=3,
map_cell_type_gene=celltype_gene_mapping,
cell_type_selected=['Lamp5 Lhx6', 'Lamp5', 'Pax6', 'Sncg', 'Vip', 'Sst Chodl', 'Sst',
'Pvalb', 'Chandelier',],
radius_step=1.5,
# cmap="Spectral_r",
# cmap="magma_r",
cmap="Reds",
gene_label_offset=0.25,
gene_label_color="black",
linkage_method="ward",
distance_metric="correlation",
# hierarchy_merge_scale=0.2,
collapse_scale=0.5,
add_cluster_node_for_single_node_cluster=False,
palette=None,
figure_size= (10, 10),
# node_shape_cell_type='o',
# node_shape_gene='s',
colorbar_width=0.01,
gene_color_min=0,
gene_color_max=None,
edge_curved=0.5,
show_figure=True,
# save='./markerDendrogram.pdf',
)
Run COSG with assigned batch_key#
In this example dataset, the cells are divided into two groups based on whther the donors are severly affected, and the information is in the Severely Affected Donor column:
[34]:
pd.crosstab(adata.obs['Severely Affected Donor'], adata.obs['Subclass'])
[34]:
| Subclass | Lamp5 Lhx6 | Lamp5 | Pax6 | Sncg | Vip | Sst Chodl | Sst | Pvalb | Chandelier | L2/3 IT | ... | L6 CT | L6b | L6 IT Car3 | L5/6 NP | Astrocyte | OPC | Oligodendrocyte | Endothelial | VLMC | Microglia-PVM |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Severely Affected Donor | |||||||||||||||||||||
| N | 285 | 598 | 134 | 312 | 1463 | 31 | 752 | 1246 | 146 | 4635 | ... | 221 | 212 | 319 | 286 | 917 | 467 | 1578 | 26 | 77 | 563 |
| Y | 20 | 55 | 6 | 17 | 88 | 0 | 28 | 53 | 11 | 191 | ... | 10 | 12 | 35 | 13 | 163 | 29 | 85 | 5 | 9 | 75 |
2 rows × 24 columns
[35]:
sc.pl.embedding(adata,
basis='X_umap',
color=['Severely Affected Donor'],
palette=self_palette2,
legend_fontoutline=2,
legend_fontsize=7,
legend_fontweight=5,
# legend_loc='on data',
# cmap='Spectral_r',
ncols=3,
size=10,
frameon=False)
We could run COSG with batch_key set as “Severely Affected Donor”, computing cosine similarities separately for each batch before averaging them. The COSG scores were then derived from these averaged similarities.
[36]:
%%time
groupby='Subclass'
cosg.cosg(adata,
key_added='cosg_batch',
# use_raw=False, layer='log1p', ## e.g., if you want to use the log1p layer in adata
mu=100,
batch_key='Severely Affected Donor', ### Set the batch key
batch_cell_number_threshold=5, ### Minimum number of cells required in a batch for a given cluster to be considered
expressed_pct=0.1,
remove_lowly_expressed=True,
n_genes_user=adata.n_vars, ### Use all the genes, to enable the calculation of transformed COSG scores
# n_genes_user=100,
groupby=groupby,
return_by_group=True,
verbosity=1
)
Finished identifying marker genes by COSG, and the results are in adata.uns['cosg_batch'].
CPU times: user 8.06 s, sys: 3.47 s, total: 11.5 s
Wall time: 11.5 s
[37]:
pd.DataFrame(adata.uns['cosg_batch']['names']).head()
[37]:
| Lamp5 Lhx6 | Lamp5 | Pax6 | Sncg | Vip | Sst Chodl | Sst | Pvalb | Chandelier | L2/3 IT | ... | L6 CT | L6b | L6 IT Car3 | L5/6 NP | Astrocyte | OPC | Oligodendrocyte | Endothelial | VLMC | Microglia-PVM | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | NKX2-1 | LINC02520 | AC006305.1 | AC004014.1 | LINC01705 | NPY | NMU | MEPE | AC006065.4 | LINC00507 | ... | CLPSL1 | AL499602.1 | SMYD1 | AC084167.1 | PRDM16 | AC004852.2 | LINC01608 | CLDN5 | COL1A2 | PTPRC |
| 1 | SFTA3 | SCNN1G | NTF3 | AC091576.1 | VIP | TMEM114 | AC113347.4 | MYO5B | AC106874.1 | LINC01484 | ... | AC073968.2 | AL357509.1 | DCSTAMP | NPSR1-AS1 | OBI1-AS1 | GPR17 | FOLH1 | TGM2 | TBX18 | SYK |
| 2 | LINC01344 | CPLX3 | AL139090.1 | AC073316.1 | AJ006995.1 | GHRH | LINC02133 | AC093765.2 | SCUBE3 | AC008574.1 | ... | AC127459.2 | RASEF | SMIM32 | LINC02543 | SLC39A12 | BEST3 | CD22 | MYCT1 | FMO2 | FYB1 |
| 3 | HCRTR2 | AC099482.2 | RELN | AL161629.1 | LINC02257 | SST | C8orf87 | LRRC38 | LINC00943 | CNGB1 | ... | AC092114.1 | LINC01654 | AC010401.1 | NPSR1 | ETNPPL | FERMT1 | MOG | CD34 | FOXD1 | IKZF1 |
| 4 | MYB | KIT | LINC01539 | LINC02408 | NPR3 | CORT | PAWR | SULF1 | NOG | LINC02714 | ... | SEMA3E | AL596257.1 | TRPC7-AS1 | CD200R1L | SLC14A1 | AC007106.1 | SH3TC2 | PCAT19 | C7 | DOCK8 |
5 rows × 24 columns
and here are the COSG results without batch_key assigned:
[38]:
pd.DataFrame(adata.uns['cosg']['names']).head()
[38]:
| Lamp5 Lhx6 | Lamp5 | Pax6 | Sncg | Vip | Sst Chodl | Sst | Pvalb | Chandelier | L2/3 IT | ... | L6 CT | L6b | L6 IT Car3 | L5/6 NP | Astrocyte | OPC | Oligodendrocyte | Endothelial | VLMC | Microglia-PVM | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | SFTA3 | LINC02520 | AC006305.1 | AC004014.1 | VIP | NPY | AC113347.4 | MEPE | AC106874.1 | LINC00507 | ... | CLPSL1 | AL499602.1 | SMYD1 | CD200R1L | ETNPPL | GPR17 | LINC01608 | CLDN5 | COL1A2 | PTPRC |
| 1 | NKX2-1 | SCNN1G | LINC01162 | AC091576.1 | CHRNA2 | TMEM114 | NMU | AC093765.2 | NOG | LINC01484 | ... | AC073968.2 | RASEF | SMIM32 | AC084167.1 | PRDM16 | BEST3 | MOG | MYCT1 | FOXD1 | FYB1 |
| 2 | LINC01344 | KIT | LINC01539 | AC073316.1 | LINC01705 | CORT | C8orf87 | MYO5B | LINC00943 | CNGB1 | ... | SMIM36 | SLITRK6 | AC010401.1 | NPSR1-AS1 | SLC39A12 | AC004852.2 | FOLH1 | TGM2 | TBX18 | SYK |
| 3 | AL355482.1 | AC099482.2 | ABCB5 | AL161629.1 | AJ006995.1 | GHRH | LINC02133 | LRRC38 | AC006065.4 | AC008574.1 | ... | KL | SCUBE1 | AC073225.1 | LINC02042 | OBI1-AS1 | LINC01546 | CD22 | SRARP | C7 | IKZF1 |
| 4 | HCRTR2 | CPLX3 | LINC01416 | AC004862.1 | NPR3 | DDC | OTOF | AC006364.1 | SCUBE3 | LINC02714 | ... | AC092114.1 | FIBCD1 | DCSTAMP | NPSR1 | SLC14A1 | VCAN-AS1 | SLCO1A2 | FENDRR | FMO2 | PIK3R5 |
5 rows × 24 columns
Save the data#
[39]:
adata.write(save_dir+'/'+prefix+'.h5ad')
[40]:
adata
[40]:
AnnData object with n_obs × n_vars = 20000 × 36601
obs: 'sample_id', 'Neurotypical reference', 'Donor ID', 'Organism', 'Brain Region', 'Sex', 'Gender', 'Age at Death', 'Race (choice=White)', 'Race (choice=Black/ African American)', 'Race (choice=Asian)', 'Race (choice=American Indian/ Alaska Native)', 'Race (choice=Native Hawaiian or Pacific Islander)', 'Race (choice=Unknown or unreported)', 'Race (choice=Other)', 'specify other race', 'Hispanic/Latino', 'Highest level of education', 'Years of education', 'PMI', 'Fresh Brain Weight', 'Brain pH', 'Overall AD neuropathological Change', 'Thal', 'Braak', 'CERAD score', 'Overall CAA Score', 'Highest Lewy Body Disease', 'Total Microinfarcts (not observed grossly)', 'Total microinfarcts in screening sections', 'Atherosclerosis', 'Arteriolosclerosis', 'LATE', 'Cognitive Status', 'Last CASI Score', 'Interval from last CASI in months', 'Last MMSE Score', 'Interval from last MMSE in months', 'Last MOCA Score', 'Interval from last MOCA in months', 'APOE Genotype', 'Primary Study Name', 'Secondary Study Name', 'NeuN positive fraction on FANS', 'RIN', 'cell_prep_type', 'facs_population_plan', 'rna_amplification', 'sample_name', 'sample_quantity_count', 'expc_cell_capture', 'method', 'pcr_cycles', 'percent_cdna_longer_than_400bp', 'rna_amplification_pass_fail', 'amplified_quantity_ng', 'load_name', 'library_prep', 'library_input_ng', 'r1_index', 'avg_size_bp', 'quantification_fmol', 'library_prep_pass_fail', 'exp_component_vendor_name', 'batch_vendor_name', 'experiment_component_failed', 'alignment', 'Genome', 'ar_id', 'bc', 'GEX_Estimated_number_of_cells', 'GEX_number_of_reads', 'GEX_sequencing_saturation', 'GEX_Mean_raw_reads_per_cell', 'GEX_Q30_bases_in_barcode', 'GEX_Q30_bases_in_read_2', 'GEX_Q30_bases_in_UMI', 'GEX_Percent_duplicates', 'GEX_Q30_bases_in_sample_index_i1', 'GEX_Q30_bases_in_sample_index_i2', 'GEX_Reads_with_TSO', 'GEX_Sequenced_read_pairs', 'GEX_Valid_UMIs', 'GEX_Valid_barcodes', 'GEX_Reads_mapped_to_genome', 'GEX_Reads_mapped_confidently_to_genome', 'GEX_Reads_mapped_confidently_to_intergenic_regions', 'GEX_Reads_mapped_confidently_to_intronic_regions', 'GEX_Reads_mapped_confidently_to_exonic_regions', 'GEX_Reads_mapped_confidently_to_transcriptome', 'GEX_Reads_mapped_antisense_to_gene', 'GEX_Fraction_of_transcriptomic_reads_in_cells', 'GEX_Total_genes_detected', 'GEX_Median_UMI_counts_per_cell', 'GEX_Median_genes_per_cell', 'Multiome_Feature_linkages_detected', 'Multiome_Linked_genes', 'Multiome_Linked_peaks', 'ATAC_Confidently_mapped_read_pairs', 'ATAC_Fraction_of_genome_in_peaks', 'ATAC_Fraction_of_high_quality_fragments_in_cells', 'ATAC_Fraction_of_high_quality_fragments_overlapping_TSS', 'ATAC_Fraction_of_high_quality_fragments_overlapping_peaks', 'ATAC_Fraction_of_transposition_events_in_peaks_in_cells', 'ATAC_Mean_raw_read_pairs_per_cell', 'ATAC_Median_high_quality_fragments_per_cell', 'ATAC_Non-nuclear_read_pairs', 'ATAC_Number_of_peaks', 'ATAC_Percent_duplicates', 'ATAC_Q30_bases_in_barcode', 'ATAC_Q30_bases_in_read_1', 'ATAC_Q30_bases_in_read_2', 'ATAC_Q30_bases_in_sample_index_i1', 'ATAC_Sequenced_read_pairs', 'ATAC_TSS_enrichment_score', 'ATAC_Unmapped_read_pairs', 'ATAC_Valid_barcodes', 'Number of mapped reads', 'Number of unmapped reads', 'Number of multimapped reads', 'Number of reads', 'Number of UMIs', 'Genes detected', 'Doublet score', 'Fraction mitochondrial UMIs', 'Used in analysis', 'Class confidence', 'Class', 'Subclass confidence', 'Subclass', 'Supertype confidence', 'Supertype (non-expanded)', 'Supertype', 'Continuous Pseudo-progression Score', 'Severely Affected Donor'
var: 'gene_ids'
uns: 'APOE4 Status_colors', 'Braak_colors', 'CERAD score_colors', 'Cognitive Status_colors', 'Great Apes Metadata', 'Highest Lewy Body Disease_colors', 'LATE_colors', 'Overall AD neuropathological Change_colors', 'Sex_colors', 'Subclass_colors', 'Supertype_colors', 'Thal_colors', 'UW Clinical Metadata', 'X_normalization', 'batch_condition', 'default_embedding', 'neighbors', 'title', 'umap', 'cosg', 'dendrogram_Subclass', 'Severely Affected Donor_colors', 'cosg_batch'
obsm: 'X_scVI', 'X_umap'
layers: 'UMIs'
obsp: 'connectivities', 'distances'
[ ]: